Primary Immunodeficiencies
- related: ID
- tags: #id
Introduction
Primary immunodeficiency disorders often present during childhood, but milder heritable forms may not manifest until adulthood. Primary immunodeficiency should be considered when patients present with frequent infections or infections with unusual organisms. The specific microbiology is often a clue to which arm of the immune system is affected (Table 38). Diagnosis of primary immunodeficiency allows directed therapy, targeted immunization, and optimized empiric treatment of secondary infections.
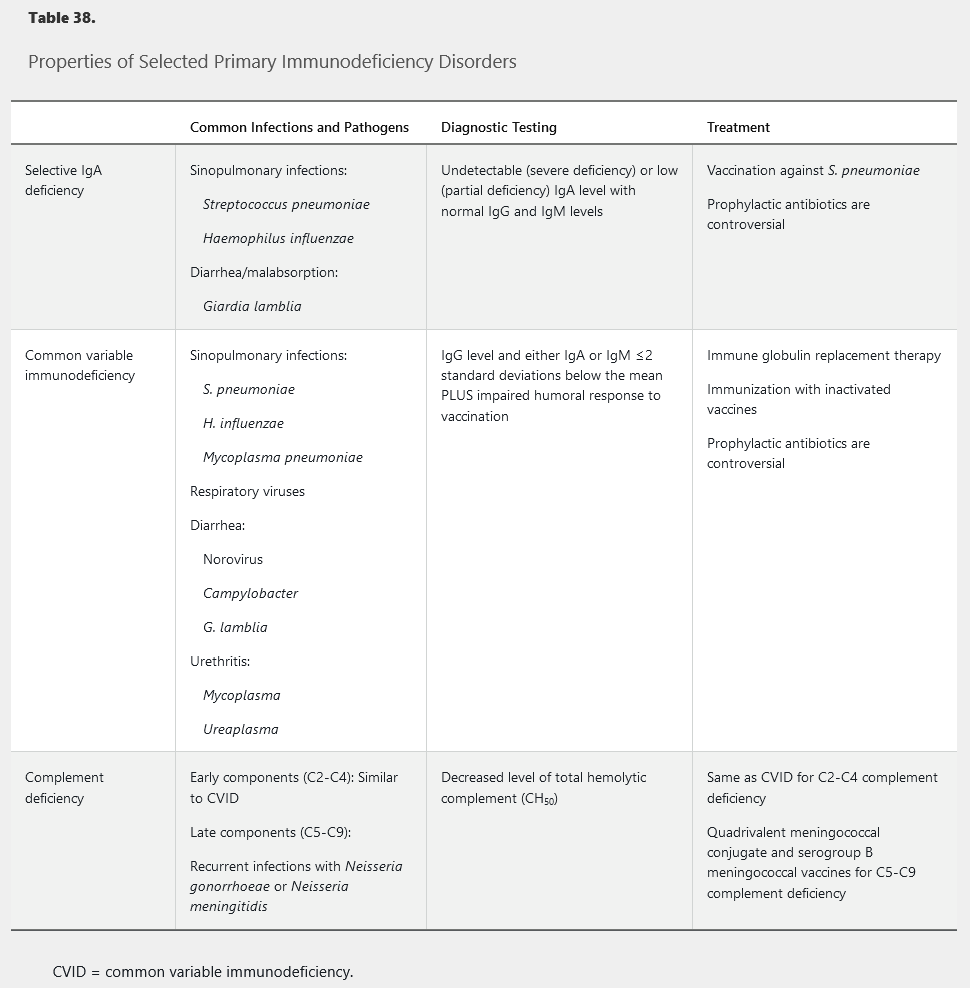
Selective IgA Deficiency
Although most patients with selective IgA deficiency (SIgAD) remain asymptomatic, it is the most common primary antibody deficiency. IgA provides mucosal immunity; therefore, patients with SIgAD are susceptible to infections of the respiratory tract and, less frequently, gastrointestinal tract. Sinopulmonary infections are the most common presenting manifestation, particularly with encapsulated bacteria (see Table 38). Anaphylaxis to blood products may occur because of the presence of anti-IgA antibodies. Testing for anti-IgA antibodies should be considered in patients with severe SIgAD or history of reaction to blood products or intravenous immune globulin infusion.
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) is a heterogenous syndrome associated with decreased quantitative immunoglobulin levels and impaired humoral response to antigens. Impaired humoral response can be tested by measuring specific antibody production before and after immunization with tetanus and pneumococcal polysaccharide vaccines. Diagnosis is usually delayed until adolescence or adulthood, although a history of recurrent infection throughout childhood may often be elicited.
CVID increases risk of infections of the upper and lower respiratory tracts caused by encapsulated bacteria, Mycoplasma species, and respiratory viruses. Gastrointestinal infections typically present with acute diarrhea caused by common enteropathogenic viruses or bacteria. Chronic diarrhea with malabsorption suggests giardiasis or chronic norovirus infection, an increasingly recognized pathogen in this population. Additionally, patients with CVID are at increased risk of autoimmune disease, inflammatory bowel disease, granulomatous disease (noncaseating granulomas in the lymphoid or solid organs), bronchiectasis, and malignancy.
Pooled immune globulin replacement therapy for CVID is performed through intravenous infusions or subcutaneous injections. Passive replacement is associated with lower rates of infections and hospitalizations. Immunization is only partially effective because of impaired vaccination response; live vaccines should be avoided. The role of prophylactic antibiotics is controversial because they can predispose patients to infection with more resistant organisms.
Abnormalities in the Complement System
Infections and other antigenic stimuli trigger an inflammatory reaction through the classical, alternative, or lectin pathway of the complement cascade system. The result is formation of the membrane attack complex, which adheres to pathogens to facilitate immune detection and destruction.
Complement deficiencies can be divided into deficiencies in early or activating components (C2, C3, C4) and late or terminal components (C5-C9). Early component deficiency, especially C4, is associated with increased rates of systemic lupus erythematosus and increased risk of infection with encapsulated organisms. Patients with early complement deficiency present similarly to patients with CVID, with recurrent sinopulmonary infections. Terminal complement protein defects lead to an inability to form the membrane attack complex and typically present with recurrent infections of Neisseria species, particularly meningococcal meningitis. N. meningitidis infection in this population tends to be less severe than in immunocompetent persons, perhaps owing to uncommon serogroups. A personal or family history of recurrent Neisseria infections is an indication to test the total hemolytic complement (CH50) level because any defect in the classical complement pathway will result in a low total level. Immunization is the mainstay of infection prevention in patients with defects in terminal complement. The Advisory Committee on Immunization Practices recommends use of a conjugate quadrivalent meningococcal vaccine as well as a vaccine active against serogroup B meningococcal infection, with a booster of the conjugate quadrivalent meningococcal vaccine given every 5 years for adults with terminal complement deficiency. These patients should also receive both pneumococcal (polysaccharide and conjugate) and Haemophilus influenzae type B vaccines.